Basic Research

The basic research done at the centre is focused on finding new genes that cause neuromuscular diseases and understanding the process of muscle degeneration leading to muscle weakness.
On the one hand, we aim to identify novel genes as the basis of our research which both informs patients and their families with these conditions and enables our ongoing research. We then aim to assess the function of these genes in various model systems with the view that understanding how loss of function in these genes causes neuromuscular disease will help us to develop novel ways to treat these devastating conditions. This process is known as translational research, where basic research develops into treatments for the patient population.
On the other hand we are very interested in understanding the process of muscle degeneration that takes place in genetic neuromuscular conditions such as Duchenne and Becker muscular dystrophy, limb girdle muscular dystrophies, facio-scapulo-humeral muscular dystrophy, spinal muscle atrophy and, Pompe disease.
The ultimate goal of our research is to identify new potential targets for therapies in these devastating disorders. For that, we employ cutting edge technology such as single nuclei/cell RNA sequencing or mass spectrometry with the aim to identify population of cells present in the muscle of patients affected and reveal their gene expression profile. We have large experience in validating our results in cell culture models, using either 2D or 3D cell models that combine satellite cells and fibroadipogenic progenitor cells obtained from the biopsies of our biobank. In parallel, the group has large experience in repurposing drugs that tackle specific pathways involved in the process of muscle degeneration and validating their effect in in vitro animal models.
Job vacancies for the research team can be found on the main University website but we are always interested in hearing from people who want to work with us
PhD Students
Current PhD students include:
-
"Design and implementation of a fully automatic pipeline for muscle MRI segmentation, fat fraction, quantification and neuromuscular disease diagnostics"

Jose Verdú-Díaz
-
"Investigating the role of the extracellular matrix in the degenerative process of skeletal muscle of patients with muscular dystrophies"

James Clark
-
"Targeting adipogenic differentiation of fibro-adipogenic progenitors in Duchenne Muscular Dystrophy"

Priyanka Mehra
-
"Exploring the molecular landscapes of neuromuscular disorders using computational methodologies"

Rasya Krishnan Gokul Nath
-
"Unravelling the pathogenesis of FSHD"

Mohi Miah
-
"High-throughput screening of antifibrotic and antiadipogenic drugs using human FAP cells"

Zoe Laidler
Our projects

Next Generation Sequencing (NGS)
The diagnosis of patients with inherited neuromuscular diseases (NMD) has changed radically in the last ten years thanks to the development and implementation in the health systems of next generation sequencing (NGS) strategies such as gene panels, exomes and more recently, genomes.
On the one hand, NGS has changed the diagnostic process reducing the number of tests required and the time to diagnosis. An earlier diagnosis results in improved patient outcome and experience, including early access to personalized management and counselling and facilitating their participation in clinical trials.
One the other, NGS also allows for basic research leading to the discovery of novel candidate genes responsible for new neuromuscular conditions so far unexplained, and in turn, a better understanding of the aetiology and pathomechanisms of other NMDs, disease modifiers and genotype‐phenotype correlations.

Ana Topf
Senior Research Associate

Leonela Luce
Research Associate

Joel Mannion
Laboratory Technician

We aim to develop a multinational multicentric diagnostic study using NGS in patients in Latin America.

This was a collaboration where we applied WES to patients with unexplained limb‐girdle weakness.

Within MYO-SEQ we identified an over-representation of patients carrying heterozygous variants…
Solve-RD was a research project funded by the EC to deliver diagnostic tests for most rare diseases by 2020.
The NeurOmics consortium generated a variety of phenotype, sequencing and other –omics data (2012-17).
Unravelling the process of muscle degeneration in genetic neuromuscular conditions
We have several projects aiming to characterize the process of muscle degeneration in genetic neuromuscular conditions such as Duchenne and Becker muscular dystrophy, limb girdle muscular dystrophies, spinal muscle atrophy and, Pompe disease.
Our goal is to understand what triggers muscle fibre death, inflammation and expansion of fibro-fatty tissue with the aim to identify new potential targets for therapies. For that, we employ cutting edge technology such as single nuclei/cell RNA sequencing or mass spectrometry with the aim to identify population of cells present in the muscle of patients affected and reveal their gene expression profile. We have large experience in validating our results in cell culture models, using either 2D or 3D cell models that combine satellite cells and fibroadipogenic progenitor cells obtained from the biopsies of our biobank. In parallel, the group has large experience in repurposing drugs that tackle specific pathways involved in the process of muscle degeneration and validating their effect in in vitro animal models.
Elucidating the role of FAP cells in the process of muscle degeneration in patients with muscular dystrophies
The overall aim of this project is to unravel the role of fibroadipogenic progenitor cells (FAP) in patients with different neuromuscular disorders. We are especially focused in Duchenne and Becker muscular dystrophy, but we have other ongoing projects in sarcoglycanopathies, Pompe disease and spinal muscle atrophy. By using advanced techniques like single nuclei RNA sequencing and spatial gene expression of skeletal muscle from patients and controls individuals we aim to identify candidate molecules that could drive FAP cell fate in these patients. Identifying and validating target molecules involved in the degeneration of the muscle can lead to find potential candidates for therapeutic approaches.
We also investigate the changes in the extracellular matrix (ECM) that occurs in muscle diseases and how the ECM differs between patients and healthy controls. A major component of muscle diseases is fibrosis which contribute to muscle chronic damage by reducing the capacity of the muscle to regenerate, maintaining chronic inflammation and reducing the response to drugs. In these projects we study how the change in the ECM modify the physical properties of the muscle, and what are the consequences of these changes. Moreover, we aim to unravel how ECM affects the molecular and cellular properties of different muscle resident cells involved in the process of muscle degeneration.
Funding: Academy of Medical Sciences Professorship Scheme and MRC.

Esther Fernández-Simón
Research Associate
James Clark
Laboratory Technical Specialist

Panos Katsikis
Laboratory Technician

Jordi Díaz Manera
Professor of Neuromuscular Disease
Underlying the pathological changes in Pompe disease and finding new therapeutical approaches
We use advanced techniques like single nuclei RNA sequencing and spatial transcriptomics, to identify the changes in cell population and gene expression profile that takes place in muscle samples of patients with Pompe disease. Identifying these pathological pathways could highlight promising candidates for therapeutic intervention.
We also investigate the effectiveness of a new therapeutic approaches in Pompe disease by optimising a novel drug delivery system. This novel delivery system aims to reduce the buildup of glycogen in muscle and brain tissue, which occurs in Pompe disease. In that sense, we evaluate the ability to reduce glycogen accumulation in vitro in cells and in vivo in animal models of Pompe disease.
Funding: Grants from Spark Therapeutics and Sanofi.
Rasya Krishnan Gokul Nath
Research Assistant
Jordi Díaz Manera
Professor of Neuromuscular Disease

Alexandra Monceau
Research Associate
Novel drugs targeting fibrosis and fatty replacement in Duchenne muscular dystrophy
The aim of this project is to evaluate potential therapeutical approaches that can modulate altered behaviour of fibroadipogenic progenitor cells (FAPs) in muscle dystrophies.
We use human samples from patients with Duchenne Muscular Dystrophy (DMD) and murine model of DMD, to test the effect of new drugs and thereby try to reduce the degenerative process of fat infiltration in muscular dystrophies.
Funding: Academy of Medical Sciences, MRC, Duchenne-UK and DiNEM PhD studentships.
Esther Fernández-Simón
Research Associate
Panos Katsikis
Laboratory Technician
Priyanka Mehra
PhD Student
Jordi Díaz Manera
Professor of Neuromuscular Disease
Zoe Laidler
PhD Student
Elucidating pathomechanisms and identifying biomarkers in Facioscapulohumeral muscular dystrophy (FSHD)
Our translational research focuses on tissue and circulating biomarkers in a peculiar form of muscular dystrophy, Facioscapulohumeral muscular dystrophy (FSHD).
Our previous studies were aimed to shed light on the meaning and value of the early inflammatory changes that precede and possibly promote muscle degeneration through still obscure but likely disease-specific mechanisms, also taking advantage from the implementation of mini-invasive techniques such as muscle microdialysis.
Our main actual goal is to fully clarify the cellular and molecular pathways driving muscle wasting in vivo in FSHD patients leveraging the sensitivity and resolution of new omic technologies applied to the study of clinically relevant patient derived samples.
Funding: Academy of Medical Sciences, MDUK and Friends of FSH Research

Sam Fitzsimmons
Project Manager
Mohi Miah
PhD student

Giorgio Tasca
Clinical Professor of Neuromuscular Science

Asmita Saha
Research Assistant Bioinformatician

Raul Fulea
Research Associate

Philipp Heher
Research Associate
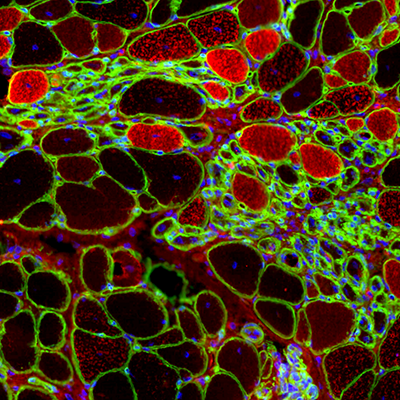
Investigating cellular and molecular changes in muscle samples of patients with Spinal Muscle Atrophy (SMA)
In this project we aim to unravel the cellular and molecular consequences of the denervation that takes place in patients with SMA produced by mutation in SMN-1 gene. For this we are using single nuclei RNA sequencing, ATAC sequencing and spatial transcriptomics.
Moreover, we aim to isolate different muscle resident cells from muscle samples of patients with SMA such as satellite cells, FAPs and glial cells and study their interaction in vitro in 2D and 3D cell models aiming to replicate the findings observed in vitro.
Funding: AFM
Rasya Krishnan Gokul Nath
Research Assistant
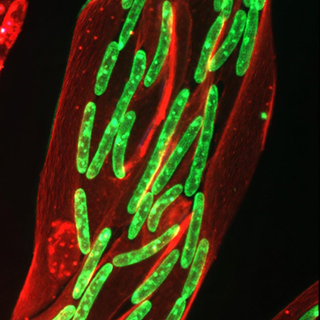
Understanding and targeting metabolic stress in Facioscapulohumeral muscular dystrophy (FSHD) and other muscular dystrophies
Muscular dystrophies are genetically diverse, yet many share common downstream features, including metabolic stress and disturbed redox homeostasis in skeletal muscle. These bioenergetic modalities are increasingly recognised as active drivers of muscle degeneration and disease progression.
Our research aims to define how altered mitochondrial metabolism and redox signalling disrupts cellular bioenergetics in FSHD and other muscle diseases. Using integrative omics coupled with functional metabolic assay pipelines in human cellular models and patients’ muscle biopsies, we aim to identify common and divergent metabolic stress pathways. Since effective redox-active therapeutic interventions require precise mechanistic insight to avoid harmful off-target effects, our work focuses on a detailed dissection of druggable metabolic pathomechanisms. This will help lay the foundation for redox therapies potentially applicable to FSHD and other muscle diseases.
Giorgio Tasca
Clinical Professor of Neuromuscular Science
Philipp Heher
Research Associate